Marine biology
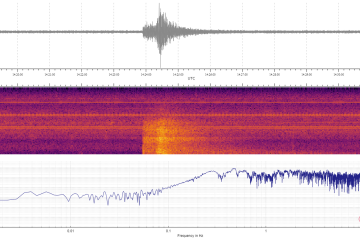
What's quieter than a fish? A school
Published April 9, 2024Swimming in schools makes fish surprisingly stealthy underwater, with a group able to sound like a single fish


Johns Hopkins is a big place. Let us make your job a little easier by connecting you with the right media representative.
Johns Hopkins University experts can provide the perspective and analysis reporters need to cover the news.