Animal cognition
'Surprisingly strategic' mice think like babies
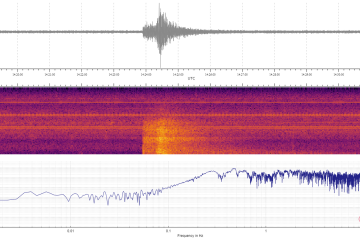
Published April 26, 2024Are mice clever enough to be strategic? Kishore Kuchibhotla, a Johns Hopkins University neuroscientist who studies learning in humans and animals, and who has long worked with mice, wondered why rodents often performed poorly in tests when they knew how to perform well. With a simple experiment, and by acting as “a little bit of a mouse psychologist,” he and his team figured it out.